







Heart failure with preserved ejection fraction (HFpEF) is a common disease with rapidly increasing incidence and prevalence due to demographic changes.1 Well-known risk factors, such as metabolic syndrome (arterial hypertension, diabetes, obesity and dyslipidaemia), chronic kidney disease and AF, predispose to the development of HFpEF, especially in older people.
Establishing the clinical diagnosis of HFpEF can be complex, as the diagnostic gold standard is the invasive evidence of elevated left ventricular (LV) filling pressures in the presence of HF symptoms and an echocardiographic preserved LV ejection fraction.2 Often, clinical symptoms of heart failure (HF) are non-specific and do not sufficiently discriminate HF and differential pathologies, and invasive work-up is unfeasible in every case. Non-invasive diagnostic algorithms, such as the Heart Failure Association’s PEFF score and the H2FPEF score, have been proposed to comprehensively associate symptoms with structural cardiac changes and elevated natriuretic peptides.2–4 However, they are characterised by limited diagnostic certainty in the detection of invasively elevated LV filling pressures and the identification of specific sub-phenotypes.5
HFpEF is associated with morbidity and a poor prognosis.6,7 However, no effective treatment addressing the whole HFpEF patient group has yet been established.2 The initially promising pharmacological approach of combined angiotensin-neprilysin inhibition failed to unequivocally demonstrate benefits.8 Current guidelines confine their recommendations to symptomatic relief and treatment of comorbidities.2 Shah et al. consequently proposed a phenotype-specific roadmap involving a multiorgan concept to provide tailored treatment strategies.9
Pathophysiological Paradigms in the Development of HFpEF
Difficulties achieving standardised management for HFpEF can be explained by the high heterogeneity of the patient group. Thorough characterisation of HFpEF patients can identify masked phenotypes, such as amyloidosis or hypertrophic cardiomyopathy, which can be partially treated.4 For HFpEF patients without an identifiable specific cardiomyopathy, a pathophysiological paradigm has been proposed by Paulus et al.10 Herein, myocardial remodelling and dysfunction result from a sequence of systemic inflammation triggered by comorbidities, especially obesity, which leads to the production of reactive oxygen species, mainly in the microcirculation, causing microvascular dysfunction.11 Subsequently, nitric oxide (NO) expression in the endothelium is reduced. In cardiomyocyte NO-triggered cyclic guanosine monophosphate (cGMP) synthesis is altered negatively, which downregulates the activity of protein kinase G (PKG).
In a rat model, PKG was shown to act like a break in hypertrophic response of the myocardium, which indicates vice versa that a downregulation of PKG results in hypertrophy and HFpEF.12 In mice, inhibition of cGMP breakdown by phosphodiesterase 5 (PDE5) prevented and reversed myocardial hypertrophy and fibrosis.13 However, in the placebo-controlled Evaluating the Effectiveness of Sildenafil at Improving Health Outcomes and Exercise Ability in People With Diastolic Heart Failure (RELAX) trial, PDE5 inhibition showed no improvements.14 An alternative pharmacological approach to enhance cGMP signalling was suggested by Lee et al.15 They found PDE9A to be upregulated, with the highest affinity and selectivity for cGMP signalling independent of the NO pathway. A PDE9A knock-out in a murine model of hypertrophic heart disease showed promising results.15 In a sheep model, Scott et al. found a beneficial effect of PDE9-inhibition compared with an untreated control, indicating a crucial role in HF and a potential therapeutic target in HFpEF.16 However, the generalisability of these findings to humans still needs to be demonstrated.
Most recently, Schiattarella et al. proposed in a murine two-hit model (metabolic and mechanical stress induced by obesity, metabolic syndrome and hypertension), whereby the abundance of myocardial NO was caused by increased activity of the inducible NO synthase (iNOS) and resulting nitrosative stress.17 This was linked to excessive protein nitrosylation within myocardial cells, including proteins central to the evolutionary conserved and cytoprotective unfolded protein response. The group elegantly demonstrated that HFpEF is characterised by a deficient unfolded protein response, which was restored after pharmacological or genetic suppression of iNOS with consecutive amelioration of an experimental HFpEF phenotype.
On a myocardial level, the common final pathway in all pathophysiological scenarios is the development of fibrosis. It has been demonstrated that clinically non-invasive phenotyping regarding the amount of myocardial fibrosis is feasible and can identify different haemodynamic abnormalities.18
However, given the proposed mechanistic link of comorbidities, inducing inflammation and the development of fibrosis, a better characterisation of the underlying inflammatory stimuli affecting the myocardium is desirable to improve diagnosis, treatment and prognosis of HFpEF, and potentially to further specify certain HFpEF subgroups in order to develop tailored therapies. Since inflammatory processes are well detected in the transcriptome, transcriptomics is supposed to be a valuable yet understudied technique to improve diagnostics and to identify new therapy targets. As Porrello et al. have pointed out, new strategies are required to explore how changes in the human transcriptome might interact with environmental stressors leading to the development of the heterogenic clinical phenotypes resulting in HFpEF.19 Moreover, new insights into the pathophysiology may enable the identification of potential new targets.
Molecular–Pathological Evaluation of Gene-expression Profile in HFpEF
Since the human genome project was successfully accomplished, identification and characterisation of gene-expression profiles and their regulating factors in diseases offer a huge advantage in understanding the pathophysiology and developing new targeted treatment strategies.20,21 The applicability of this approach has been demonstrated in oncology, where gene-expression profiling of tumour tissue has been used to individualise treatments and consecutively enhance survival and medical quality in the past few decades. As a prominent example, the treatment of breast cancer has been revolutionised by analysing the individual tumour gene expression.22 In a 5-year follow-up, Druker et al. described promising results in patients suffering from chronic myeloid leukaemia treated with a specific antibody therapy (imatinib) adapted to the underlying gene mutation (BRC-ABL tyrosine-kinase).23
At the molecular level, gene expression produces messenger RNA (mRNA) during the process of transcription, which forms the basis for protein synthesis during translation. Gene expression itself can be regulated by the transcribed, but not protein-translated, non-coding RNA, for which several sub-types are known. Among them, microRNA (miRNA, 20–25 nucleotides) and long non-coding RNA (lncRNA, >200 nucleotides) are the most comprehensively investigated, due to their high regulatory potential. The miRNAs are one of the major regulatory gene families inducing degradation or repression of mRNA through specific base pairing. Their small size allows them to target many genes, interacting with other miRNAs to create complex regulatory networks, which affect cellular processes, such as cell differentiation. Therefore, they have become attractive targets for biomarker studies and may potentially be used as treatment targets in the future.24,25 The lncRNAs act as transcriptional factors and are more tissue specific than mRNA. Generally, lncRNAs have been less well-studied in human pathophysiology, because of methodological limitations in the past. However, newer sequencing methods, such as next-generation sequencing (NGS), have overcome some of those limitations and pose an attractive tool for further investigation in human diseases.26,27 While non-coding RNA analysis may have great potential for biomarker establishment, the investigation of its regulative function is complex. Analysing protein-coding mRNA reflects the definitely transcribed proteins in the analysed tissue as a result of transcription and regulatory processing.
A number of techniques exist for transcriptomic analysis. Three commonly applied approaches are real-time quantitative polymerase chain reaction (RT-PCR), microarray analysis and NGS. In RT-PCR amplification of RNA is monitored in real time and exact amounts of amplified RNA can be quantified. It has a low dynamic range and is precise. However, this method is limited by a low scalability and has virtually no power to discover potentially novel transcripts.28 The microarray technique overcomes the scalability limitation, allowing for transcriptome-wide analysis. Microarrays use probes to simultaneously analyse the expression of thousands of genes, with each probe targeting a unique sequence within a given mRNA transcript of interest. The result is a snapshot of actively expressed genes at a given point in time.29 However, relying on predefined probes hinders the detection of previously unidentified genes or transcripts. NGS is the most recent technique and combines a high dynamic range, high precision and high throughput with the ability to detect novel transcripts, but it is the most expensive option.30
Gene-expression Analyses in Clinical Trials
The potential of transcriptomic tissue analysis in cardiology was demonstrated by Heidecker et al. who published 100% specificity and sensitivity in detecting myocarditis in myocardial biopsies from a mixed dilated (n=32) and inflammatory (n=16) cardiomyopathy cohort by a transcriptome-based biomarker containing 62 out of 9,878 differentially expressed genes on microarray and RT-PCR assessment.31 In HFrEF, identification of several monogenic subtypes has been suggested.32 Furthermore a reactivation of fetal genes in the adult failing heart has been described based on a complex process involving transcriptional, post-transcriptional and epigenetic regulation of the cardiac genome.33 However, little is known about potential genetic determinants of HFpEF.
Given the diagnostic potential of transcriptome analysis, gene expression could also facilitate the differentiation of HFpEF patients.4 Indeed, transcriptomic analysis of mRNA using an NGS approach of myocardial biopsies of HFpEF patients (n=5) and non-HF patients (n=11) taken during coronary artery bypass grafting showed up to 750 differentially expressed genes in the myocardium related to impaired myocardial contraction, tissue remodelling, extracellular matrix organisation and oxidative phosphorylation.34 Those mechanisms are known to result in systolic dysfunction, especially of the LV, which has also been suggested as a characteristic of HFpEF patients.35 However, uneven distribution of relative myocardial ischaemia in both groups cannot be ruled out as a confounder.
Apart from these studies, human myocardial transcriptomics in HFpEF have rarely been described, due to the fact that harvesting myocardial tissue is an invasive procedure and currently not routinely performed in patients with preserved ejection fraction. This means that the potential benefits of analysing myocardial tissue in HFpEF might largely stay limited to clinical studies. Thus, we need to find a potent diagnostic tool, potentially using blood biomarkers, that could reasonably be applied more universally in routine clinic tests.
Circulating RNA markers, mainly miRNAs that can be assessed conveniently in the peripheral blood, could be the answer. Ellis et al. evaluated circulating miRNA expression in plasma using a RT-PCR approach in patients admitted to hospital for acute shortness of breath showing a distinct gene-expression profile depending on underlying HF (HFpEF and HFrEF, n=32) or chronic obstructive pulmonary disease (COPD, n=15) compared with the control group (n=14). After external validation (n=150), they found that of 742 evaluated miRNAs, four could distinguish between the clinical scenarios, with a diagnostic value mainly in combination with the well-established cardiac biomarker N-terminal pro B-type natriuretic peptide (NT-proBNP) supporting the diagnostic potential of miRNAs in the blood plasma.36 While the study could not validate the use of miRNAs to distinguish between HFrEF and HFpEF, discrimination between HFrEF (n=15; n=39) and HFpEF (n=15; n=19) was shown to be possible by analysing circulating miRNA in plasma in two other studies using an RT-PCR and microarray approach, respectively.37,38
More challenging, but of high relevance in daily practice, is the differentiation of a well-compensated, non-acute HFpEF patient from a non-HF patient. Wong et al. addressed this issue in about 900 compensated HF patients and 800 non-HF patients by analysing eight miRNAs in plasma using an RT-PCR approach. Multiple miRNA panels in combination with NT-proBNP were shown to have a diagnostic power in identifying non-acute HF and discriminating between HF phenotypes. Results were independently validated in an external cohort.39
Peripheral Blood Mononuclear Cells as a Potential Peripheral Diagnostic Biomarker
Peripheral blood mononuclear cells (PBMCs) are interesting in this context, as they are peripherally available, nucleus-carrying cells and have the potential for full transcriptome analysis.40 Additionally, they are integral to systemic inflammation, which is proposed to be the common underlying condition, as described above.
Gerling et al. investigated the role of PBMCs in hyperaldosteronism and hypertensive heart disease in rats using a microarray approach. Transcriptional signatures in PBMCs showed comparable results to the cardiomyocytes. Whether PBMCs may serve as early and non-invasive sentinels for myocardial gene expression in humans needs to be further evaluated, but in any case the PBMC transcriptome might inform on gene regulation in the inflammatory state, which is assumed to be the cornerstone of HFpEF pathophysiology.41
Evidence for a pathology-specific PBMC transcriptome comes from Gupta et al., who found a specific signature of PBMCs in patients with dilated cardiomyopathy (DCM, n=44) resulting in HFrEF compared with healthy controls (n=48) using a microarray approach. External validation was performed in patients with breast cancer as an extracardiac disease, which showed a distinct gene expression comparable to healthy controls.42,43
In patients with HFpEF, a study has shown a positive correlation between exercise capability on cardiopulmonary exercise testing and expression of microRNA-208b in PBMCs (n=56) using an RT-PCR approach. These results indicate the possibility to measure the symptom-correlated severity of HFpEF in PBMCs.44
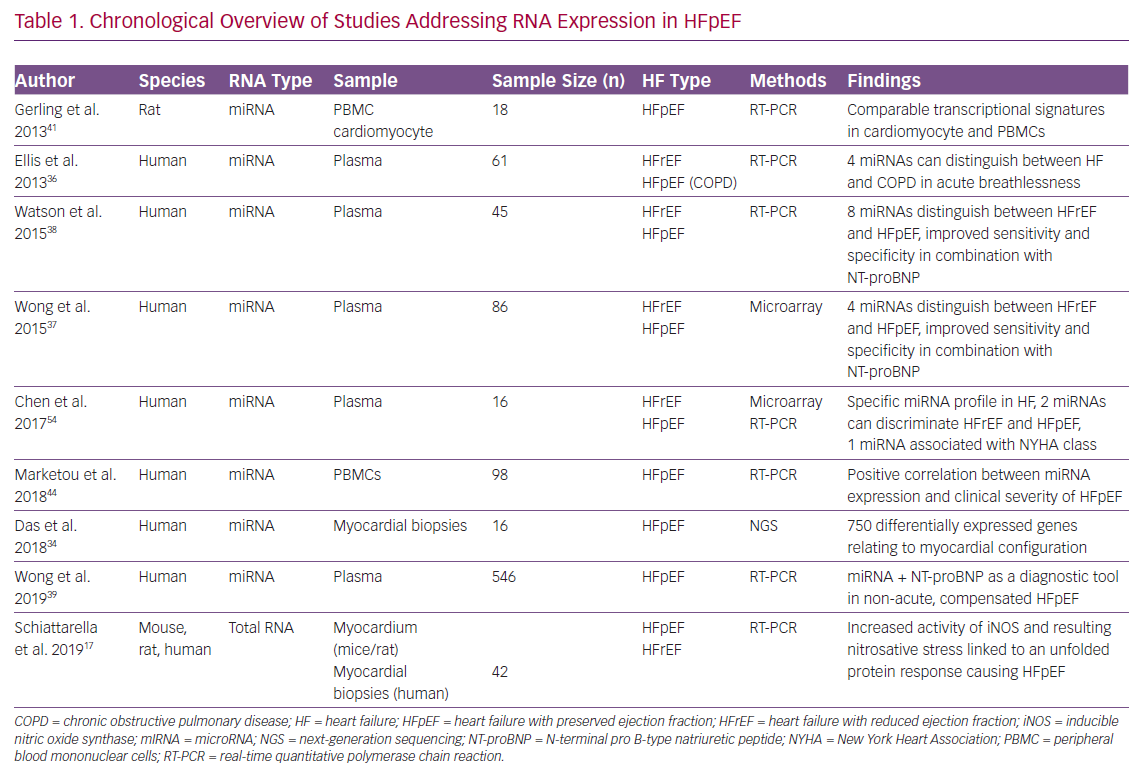
A chronologically structured overview of currently published studies addressing RNA analysis in HFpEF conditions is given in Table 1 comparing the considered RNA species, sample types, study sizes, HF types and the main findings.
Translation of Transcriptomic Discoveries to Bedside
Given the heterogeneity of the HFpEF syndrome, application of one therapeutic agent addressing all HFpEF patients appears unlikely. In contrast, better characterisation of patients in subgroups with predominate phenotypes might help to personalise pharmacological treatment.9 Transcriptomic research and genetic phenotyping has the potential to unravel individual HFpEF pathomechanisms and to identify key treatments for new therapies in the future. Moreover, clustering patients according to transcriptomic findings might offer a more immediate clinical benefit. While the pathophysiological paradigm focuses on a myocardial inflammation triggered myocardial (and peripheral tissue) remodelling in HFpEF, which results in a downstream fibrotic effect, clinically patients present as a fibrotic or inflammatory phenotype.18
The potential of transcriptomic studies to be used as complex biomarkers could help the clinician to tailor treatments with already available pharmacological agents. For example, anti-fibrotic treatments with spironolactone failed to prove overall benefit in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) study, but might be a useful drug in the presence of enhanced myocardial fibrosis and stiffness, as indicated by a beneficial effect in patients with less atrial compliance (and more BNP).45–47 Recently, the use of specific anti-inflammatory therapies, such as interleukin-1beta and low-dose colchicine, have been shown to offer benefit in cardiovascular disease with chronic inflammation, and might further complement the clinical toolbox of heart failure treatments if used in susceptible patients.48,49
Differential Gene Expression in Cross-sectional and Longitudinal Analyses
Out of the Framingham Heart study (n=8,372), which was established in 1947, Andersson et al. recently published cohorts of prevalent HFrEF (n=62) and HFpEF (n=35) to elucidate potential genetic contributors to cardiac remodelling and HF.50 Genome-wide single-nucleotide polymorphisms, gene expression and DNA methylation were characterised using a transomics analytical approach. During a mean follow-up of 8.5 years, 2.7% (n=223) and 2.8% (n=234) developed HFrEF and HFpEF, respectively. Gene-expression analysis found distinct profiles in prevalent and incident HFrEF, as well as HFpEF indicating molecular contributors of HF development. However, the findings need to be validated externally.
Heart Failure Cohort of LIFE-Heart Study
At the Heart Center, Leipzig, and Leipzig University, the Leipzig Heart study (LIFE-Heart) recruited 6,995 patients with suspected coronary artery disease (CAD), stable CAD or acute MI from 2006 to 2014. Patients were characterised by clinical data, echocardiography, coronary angiography and laboratory and molecular genetic tests.51 Samples were also stored in biobanks, allowing further retrospective analyses. From this cohort, we identified 719 patients fulfilling HFpEF diagnostic criteria and 1,106 patients without HF and available gene-expression data. At baseline, gene-expression profile of PBMCs in HFpEF and non-HF samples was performed using a microarray approach. In this huge cohort, we observed a HFpEF-specific gene-expression profile compared with non-HF patients.52,53 External validation in an appropriate and independent cohort confirmed these findings.
In addition, a prospective follow-up study of patients initially not affected by HF was established to detect the incidence of HFpEF in a longitudinal assessment (Predictors for the Development of Heart Failure and preserved Ejection fraction; PREDICT-HFpEF). Genome, proteome and PBMC transcriptome are characterised at baseline and after an average follow-up period of 7 years.
Based on this and other large-scale cohorts, the gap between cross-sectional and longitudinal studies in HFpEF could be closed. Our multi-omics approach has the potential to improve identification and characterisation of underlying molecular mechanisms causing systemic inflammation conditions in these patients and their impact on HFpEF development. Finally, biomarkers and classifiers developed in a cross-sectional approach in order to distinguish HFpEF from other entities could be longitudinally validated by our approach.
Conclusion
Transcriptome analysis appears to be a promising tool for the identification and characterisation of HFpEF patients. Multi-omics analysis in HFpEF could enhance our understanding of underlying pathophysiologies and may help to identify patients at risk. It also offers the hope of new potential therapeutic targets.